Accessing CVA Portal
How can I access CVA Portal?
CVA Portal can be accessed by NHS users on the HSCN network here: https://cva.genomicsengland.nhs.uk
You should use your Genomics England username and password.
If you cannot access CVA please contact the Genomics England Service Desk
Search
How do I search for a particular case in CVA?
The search bar on the home page and at the top of all CVA pages can be used to search for cases. You can search for Genomics England Patient ID, Family ID or Case ID (Interpretation Request ID).
When searching for a Case ID (Interpretation Request ID) you do not need to include the CIP prefix or request version as CVA stores the most recent
e.g. if searching for SAP-1234-1 use 1234.
How do I search for a particular Panel, Clinical Indication or Phenotype?
Using the CVA search bar you can search for HGNC gene symbols, Ensembl Gene IDs (ENSG), Clinical Indications (aka recruited disorders), HPO terms and HPO IDs.
How do I search for a particular Gene?
Use approved HGNC symbols in the CVA search bar.
When searching for HGNC gene symbols CVA will return cases that have that gene highlighted by interpretation services applied to the case
If you are interested in only cases where the gene has been included in the reporting outcomes questionnaire you can further filter the search results using the "reported gene" filter
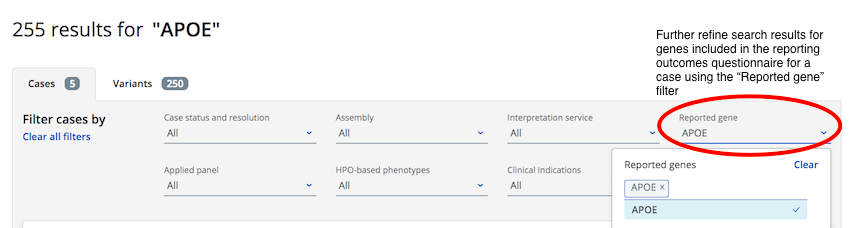
Cases
How do I know if my case has a pathogenic variant?
Users complete the reporting outcomes questionnaire in the Interpretation Portal and record whether the case is either solved, partially solved, unsolved or unknown
The answer to this question determines what is displayed in the primary findings tool tip

Search results can be filtered using the "Case status and resolution" filter
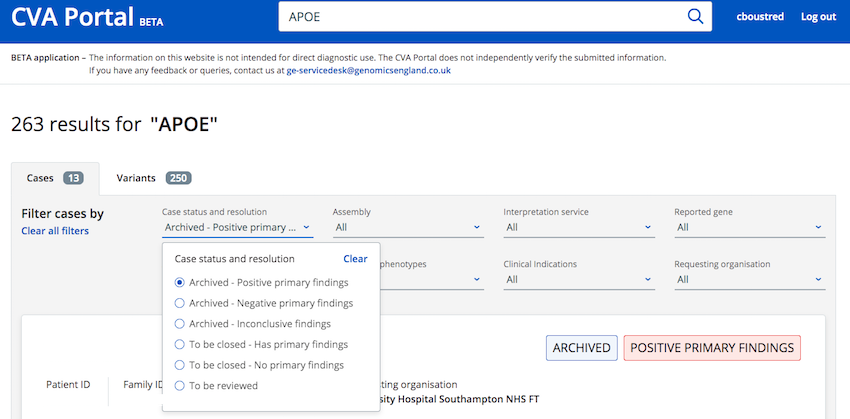
By opening the case view for a case with positive primary findings users can identify the primary findings in the case by reviewing the Summary of Findings in the side bar
NOTE: it is possible cases are marked as solved or partially solved in the outcomes questionnaire without having any variants flagged in the outcomes questionnaire
How do I know if my variant has been seen in other cases before?
On the variant list of the "Case Page" there is a column, "Cases with the variant". Click on the variant co-ordinates link to take you to a "Variant Page" that provides further details of the variant and the cases that have it
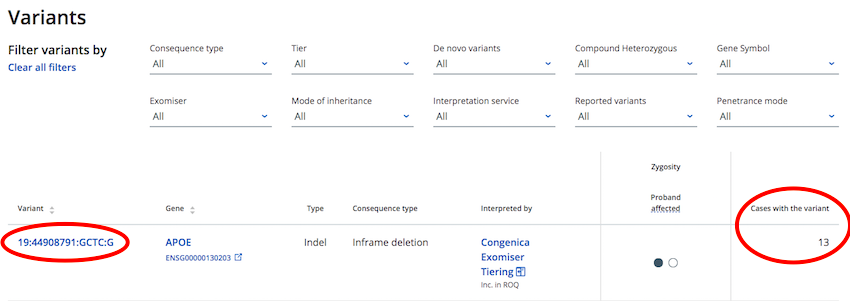
From any CVA Portal variant page, scroll to bottom of the page to view a lists of all cases that have that same variant.
From a Case View page the variant table has a column that shows a count of other cases that have this variant included in the Primary Findings. Clicking through to the variant page provides details of those case.
How do I view similar cases to mine?
From the CVA Portal Case Viewer page select "View Phenotypically Similar Cases" to open a side bar that lists phenotypically similar cases.
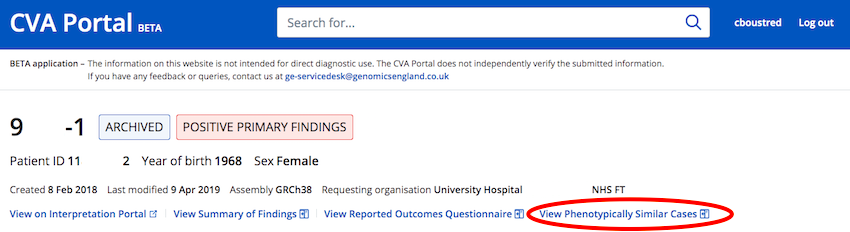
How is case similarity determined?
Phenotypic case similarity score follows a similar approach to Phenodigm (Smedley 2013), although it uses the Lin distance based on the Most Informative Common Ancestor (MICA) and HPO annotations over OMIM to calculate the information content of HPO terms.
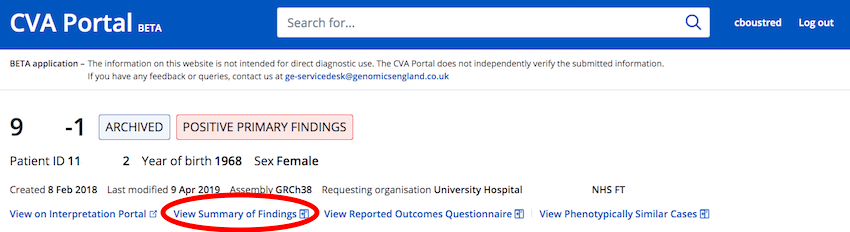
How do I see the Summary of Findings for my case?
For cases that have had a Summary of Findings generated in the Interpretation Portal (or CIP /DSS) there is a "View Summary Of Findings" button at the top of any CVA Portal case viewer page.
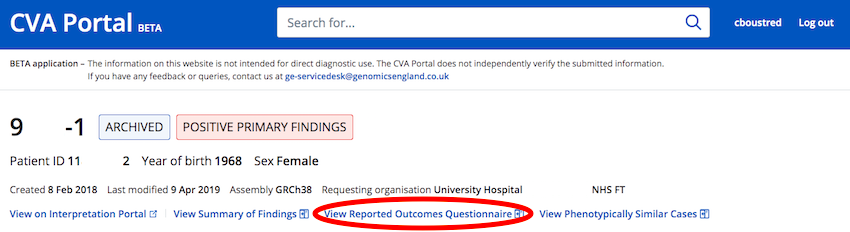
How do I see the Reporting Outcomes Questionnaire for my case?
For cases that have had a Reporting Outcomes Questionnaire generated in the Interpretation Portal there is a "View Summary Of Findings" button at the top of any CVA Portal case viewer page.
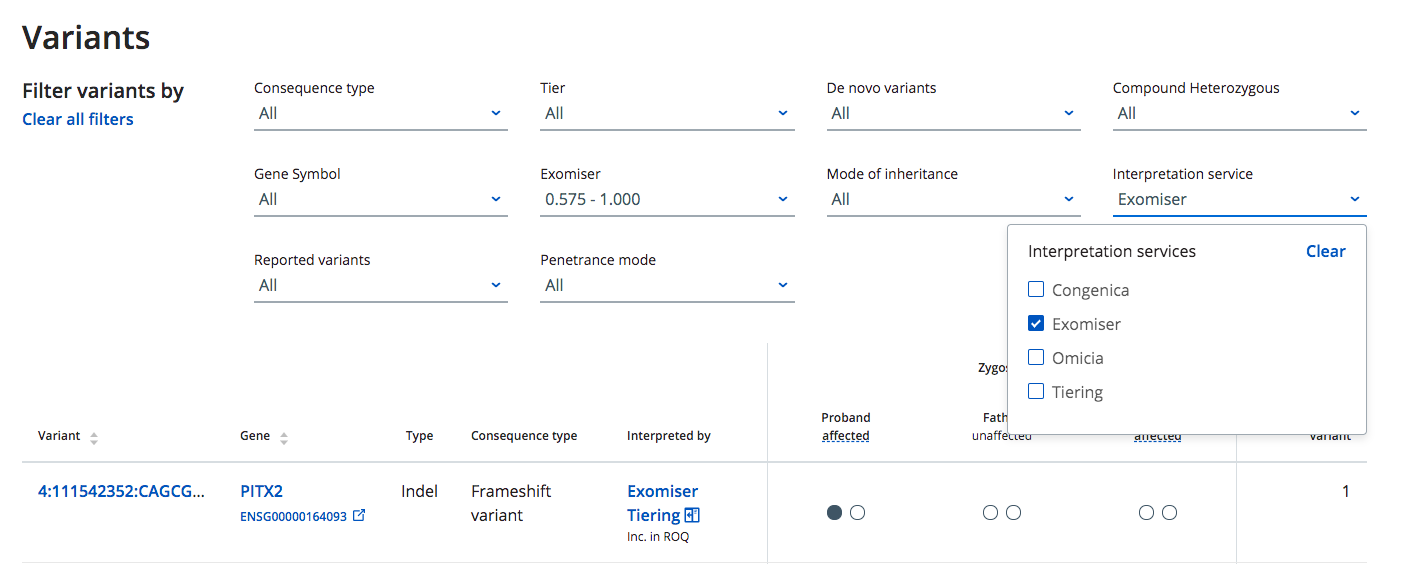
How do I filter variants in my case based on the Interpretation Service?
Lists of variants highlighted by the Interpretation Services (e.g. Tiering & Exomiser) applied to your case can be seen on the case page
The "Interpretation Service" filter allows you to filter variants identified by different Interpretation Services applied to your case.
If an Interpretation Service filter option is not displayed it means, either, that Interpretation Service has not been applied to the case or the results for that Interpretation Service have not yet registered in the CVA database
NOTE: The Exomiser score filter is present on the case page by default and will not work if the Exomiser Interpretation Service has not been applied to your case. Please check the Interpretation Service filter first to determine whether there are variants from Exomiser present in your case
How do I filter for only cases that have a Positive Diagnosis?
Search results can be filtered using the "Case status and resolution" filter
Positive primary findings
At least one variant is reported as 'pathogenic' or 'likely pathogenic'
Negative primary findings
All variants in the primary findings are 'benign' or 'likely benign' OR at least one variant is reported as 'variant of unknown significance' and other variants are 'benign' or 'likely benign' OR no variants were included in primary findings
Inconclusive primary findings
All variants in the primary findings are 'variant of unknown significance'
Variants
How is HGVS calculated in CVA Portal?
HGVS annotation in CVA Portal is obtained from CellBase which provides HGVS according to the recommendations here: https://varnomen.hgvs.org/recommendations/general/
How are transcripts selected HGVS annotation?
HGVS on the variant view page in CVA Portal can be seen for all transcripts by clicking on the side bar icon. On the main variant view page, the default HGVS transcript is selected based on the following rules:
- The transcript must be "protein coding" - if more than one transcript satisfies this rule then...
- The transcript must have an Ensembl "gencode basic" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with an "CCDS" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with the longest coding sequence length - if more than one transcript satisfies this rule then...
- The transcript with the largest number of exons is selected
If there is still more than one transcript for a gene then one is selected at random
How do I find a list of all variants classed as Pathogenic or Likely Pathogenic?
This is currently not possible in the CVA Portal. It is a feature we wish to add in 2019.
How do I see a list of all Tier 1 variants?
This is currently not possible in the CVA Portal. It is a feature we wish to add in 2019.
Genes
How are transcripts selected for the Gene View Page?
A single transcript was selected for determining the consequence types displayed for a gene based on the following rules:
- The transcript must be "protein coding" - if more than one transcript satisfies this rule then...
- The transcript must have an Ensembl "gencode basic" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with an "CCDS" annotation flag - if more than one transcript satisfies this rule then...
- The transcript with the longest coding sequence length - if more than one transcript satisfies this rule then...
- The transcript with the largest number of exons is selected
If there is still more than one transcript for a gene then one is selected at random
How do I see a list of all loss of function variants in my gene?
From the "Variants" tab after performing a search you can filter variants using the "Consequence Type" filter
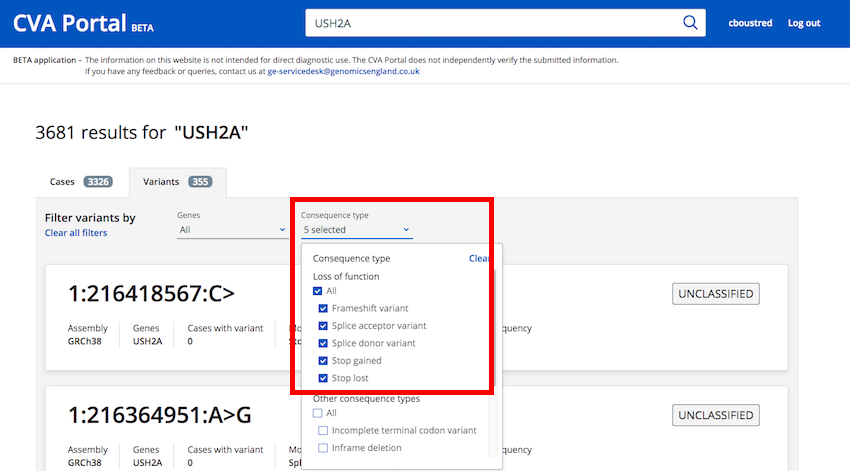
Loss of Function consequence types have been grouped into a single selection option
How do I see all classified variants in my gene?
The Gene Page can accessed here:
cva.genomicsengland.nhs.uk/gene/[HGNC symbol or ENSG]
e.g. https://cva.genomicsengland.nhs.uk/gene/APOE or https://cva.genomicsengland.nhs.uk/ENSG00000130203
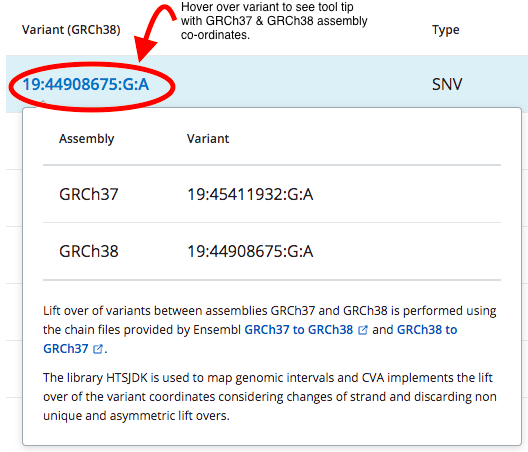
How do I know which genome assembly is relevant to my case?
All variants in the CVA database are lifted over to GRCh38, however, if you case was originally interpreted on GRCh37 these co-ordinates will be displayed.
You can "hover" over any variant co-ordinate in CVA Portal will show you the co-ordinates in both GRCh37 and GRCh38
Variant Interpretation Logs (VILs)
What are VILs?
VILs aim to capture the decision making process that leads to a variant classification. They record ACMG scoring, comments and classifcations added to variants in the Decision Support Systems e.g. Congenica
When a user completes their interpretation of a case in the DSS (e.g. in Congenica they would click the "Review Complete" button) the VIL data associated with that case is automatically pushed to CVA via the CIP-API
Where can I see VILs in CVA?
Currently VILs are shown on the case pages of CVA. There will be a side bar under any variant listed on the case page that has a VIL
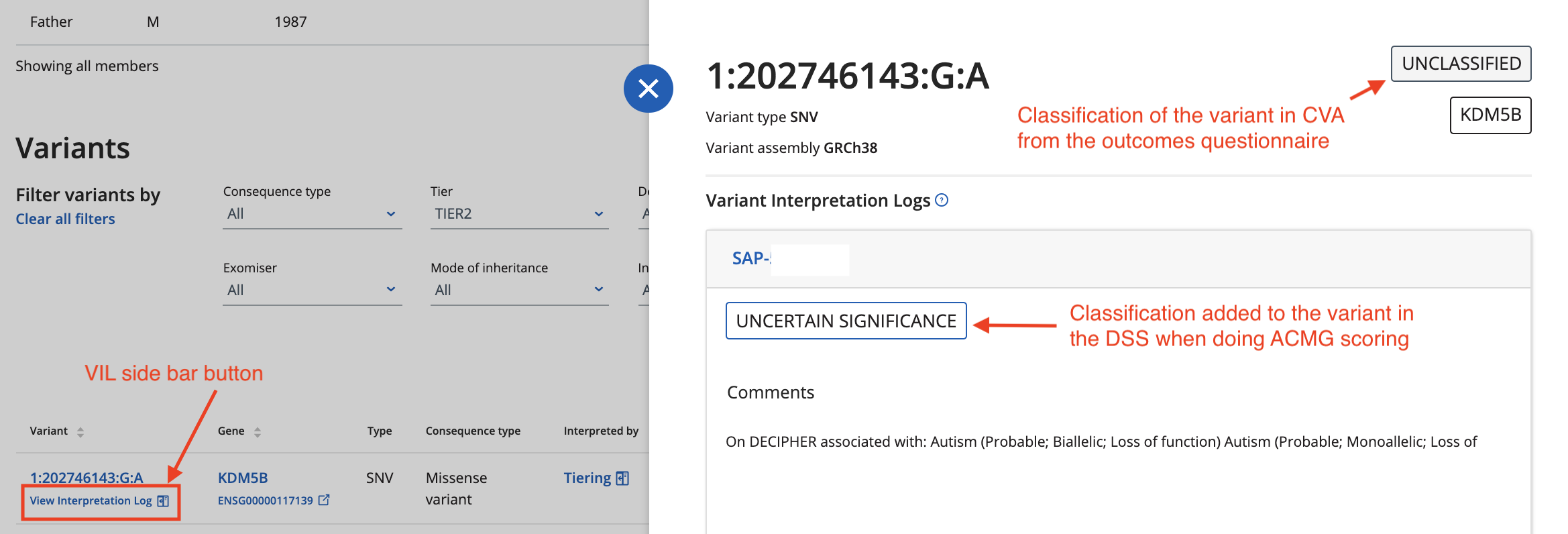
In the future we hope to make VILs available in other pages in CVA
API
How do I programmatically access data in CVA?
Data in CVA can be accessed through a REST-API.
There is a python client in PyPi.
Labs
How do I contact a lab about a case?
All cases in CVA portal list the laboratory who requested the test and where available details the person from that lab who created the Summary of Findings and the Reporting Outcomes Questionnaire.
If you'd like to contact a laboratory about a case or variant you have seen in the CVA Portal you can look up their details.
A complete listing of laboratories in the new GMS should become available in 2019.
How do I see only cases from my GMC / GLH?
Search results can be filtered by "requesting organisation" to filter only results from a particular GMC / GLH.